JaSMInJapan Registration System for Metabolic & Inherited Diseases


副腎白質ジストロフィー(ALD)
岐阜大学 生命科学総合研究支援センター ゲノム研究分野
下澤 伸行
副腎白質ジストロフィー(adrenoleukodystrophy、ALD)は脳の白質と副腎に異常をきたす遺伝病で、幼児期から成人にかけて、色々な症状で発症します。大脳型の場合には、唯一の治療法が発症早期の移植であることより、できるだけ早期に診断することが予後の改善に重要です。また、ALDは厚生労働省の指定難病にも認定されています。
1.副腎白質ジストロフィーはどんな病気ですか?
脳や脊髄における脱髄(神経線維を覆っている髄鞘と呼ばれる鞘の部分の崩壊)と副腎の機能不全を特徴とする遺伝病(病気の発症に遺伝子が関わる病気)です。患者さんの多くは男性で、様々な病気のタイプがあります。女性でも年齢とともに歩行障害等の症状をきたす方がいます。全身の組織や血液に極長鎖脂肪酸が蓄積しており、血中の極長鎖脂肪酸を測定することより診断が可能になります。
2.国内にどのくらいの患者さんがいますか?
2012年度におけるALDの国内難病医療受給者証保持者は193人でしたが、2016年度に厚生労働省の研究班で行った全国調査の結果では400人程度の国内患者の存在が推測されています。
3.どのような原因で発症しますか?
X染色体に存在するALDの原因遺伝子(ABCD1)の変異により起こるX連鎖性の遺伝病です。ABCD1遺伝子が作るタンパク質は、極長鎖脂肪酸を細胞内にあるペルオキシソーム内に輸送する機能があると考えられています。極長鎖脂肪酸はペルオキシソームの中で分解されて利用されるのですが、ABCD1遺伝子に変異があると正常なタンパク質が作られず、極長鎖脂肪酸が輸送されずに細胞内に蓄積すると考えられています。ただ、ABCD1遺伝子の変異や極長鎖脂肪酸の蓄積がどのようにして、 ALDを発症するかについての詳しい機序は必ずしも明らかにされていません。

4.遺伝について教えてください。
染色体は通常、常染色体として22対、44本に加えて、性染色体として女性はX染色体を2本、男性はX染色体とY染色体を1本ずつ、併せて男女共に46本の染色体を持っています。ALDの病因遺伝子はX染色体上にあり、男性ではX染色体が1本しかないため、その遺伝子に変異があると重症なタイプも含めて発症します。一方、女性ではX染色体が2本あるので、片方のABCD1遺伝子に変異がある方を保因者と呼び、発症しない、もしくは発症しても軽症になる傾向があります。女性保因者のお子さんは、男児の場合には50%で患者、女児の場合には50%で保因者になると考えられます。一方、男性患者のお子さんは男児にはX染色体は伝わらないのでALDになる可能性はなく、女児には必ず伝わりますので、皆、保因者になると考えられます(図1)。
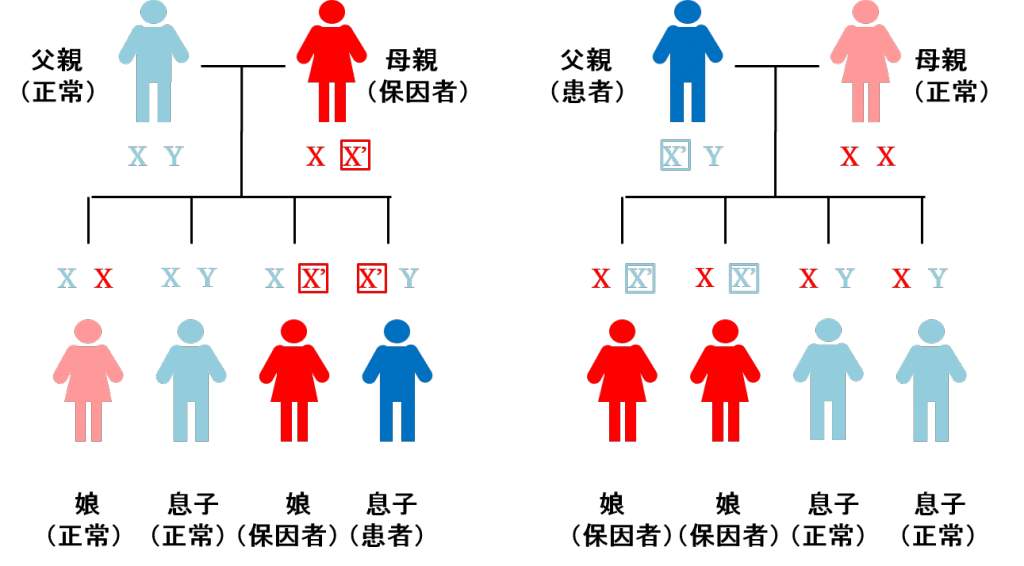
図1 ALDの遺伝
5.どんな病気のタイプがありますか?
多くの病気のタイプ(病型)があり、発症した年齢や症状で分類されています。また、ABCD1遺伝子変異の種類と病気のタイプとの関連は明らかではなく、同じ家系内や兄弟の患者でも異なる病型で発症することがあります。
【小児大脳型】2~10歳に性格や行動の変化、視力や聴力の異常、知能障害、歩行障害、けいれんなど、様々な症状で発症して、数年で寝たきりになることもあります。
【思春期大脳型】11~21歳に小児大脳型と同じような症状で発症しますが、進行はやや緩やかな場合もあります。
【副腎脊髄ニューロパチー(AMN)】10代後半~成人にかけて歩行障害がゆっくりと現れ、排尿障害などが加わります。進行は緩やかですが、大脳型に移行することがあります。
【成人大脳型】性格変化や精神症状、認知症を疑わせる症状で発症し、比較的急速に進行する場合もあります。
【小脳・脳幹型】ふらつき歩行や呂律が回らないなどの症状で発症し、大脳型に移行することもあります。
【アジソン型】2歳以降から成人期にかけて色素沈着や嘔吐、食欲不振、無気力などの副腎不全症状で発症します。発熱やストレスを契機に急激な症状で発症することもあり、注意を要します。また、大脳型やAMNに移行することがあります。
【女性発症者】女性保因者の一部に加齢とともにAMNに似た症状で発症することがあります。
【発症前男性】患者さんからの家系解析等で診断されることがあり、どの病型になるかは判りませんが、大脳型や副腎不全の発症に早くから対応するために発症前診断は推奨されています。
6.具体的にどのような症状で疑えばいいですか?

小児や思春期大脳型では、斜視や、「物を見づらそうにしている」ことから眼科を受診しても視力検査で異常を認めなかったり、眼鏡で矯正できなかったケースや、「聞き返しが多くなった」ことから耳鼻科を受診しても聴力検査で異常を認めなかったケース、また学校で「落ち着きがなくなってきた」、「おかしな行動をするようになった」「成績が落ちてきた」「書き方や話し方がおかしくなってきた」などの症状をきたすケースがみられます。
成人大脳型では、「怒りやすくなった」などの性格の変化や「仕事のミスが多くなった」「家庭内のトラブル」などがみられることがあります。
AMNでは、足がつっぱる、歩きにくい、転びやすい、その後、尿や便が漏れやすいなどの症状が、小脳・脳幹型では、呂律が回りにくい、ふらつくなどの症状をきたして、いずれも脊髄小脳変性症の鑑別の中で診断されるケースもみられます。
アジソン型では、食欲不振、無気力、体重減少や全身の色素沈着(爪床や歯肉、口唇、外陰部にも注意)などの副腎不全症状をきたしますが、ストレスや感染時に嘔吐や意識障害などの急性症状をきたす場合もあります。また、既に慢性副腎不全として治療中の男性患者の中で診断される方もみられます。
7.女性保因者はどのような経過で発症しますか?
女性保因者では、成人以前の発症はほとんどありませんが、歳をとるとともに臀部から腰や下肢の痛みや一部には歩行障害に至る方もみられます。また、尿や便の失禁がみられることもあります。一方で、大脳型や副腎機能不全を起こすケースは極めて稀です。
8.どのようにして診断されますか?
大脳型では、脳MRI検査にて疑われて、血中極長鎖脂肪酸の増加にて診断されます。AMNや脳幹・小脳型、アジソン型を含めて男性患者では、症状に極長鎖脂肪酸の蓄積がみられればほぼ診断され、ABCD1遺伝子変異を認めればより確実です。一方、女性保因者では、発症の有無に関わらず極長鎖脂肪酸が増加するのは80%程度とされており、遺伝子解析が診断に際し、重要になります。
岐阜大学ゲノム研究分野では、ALDが疑われる患者さんについては極長鎖脂肪酸とABCD1遺伝子解析を無償で提供しています(岐阜大学ALD&ペルオキシソーム病ホームページ)。
9.治療法はありますか?
男性患者の副腎不全に関しては、アジソン型に限らず大脳型やAMNでも70~80%に副腎機能低下を認めるため、定期的な副腎機能評価と適切なステロイド補充療法が必要です。これは移植後の患者さんについても同様です。一方、女性保因者では発症例も含めて副腎機能不全は極めて稀です。
大脳型に関しては、唯一の治療法が造血幹細胞移植で、発症早期ほど予後の改善が期待されます。ドナーとしてはHLA一致の同胞(患者でも保因者でもない)が最も推奨されています。近年は、臍帯血バンクの整備や移植技術の向上により、骨髄非破壊的前処置による臍帯血移植で、移植リスクや準備期間が改善され、良好な治療成績を挙げています。一方で、進行した症例では予後改善は必ずしも期待されず、早期の診断が極めて重要になります。
ロレンツォオイルの投与は、血中の飽和極長鎖脂肪酸を低下させるものの、発症後の大脳型やAMN患者の予後改善は期待されません。ただ近年、発症前投与により大脳型発症のリスクを低下させる可能性が検討されています。
10.発症前に診断することは必要ですか?
男性患者の場合、副腎不全や大脳型発症に対して早期に介入することにより予後改善が期待されるため、発症前診断は推奨されています。その場合、副腎不全や小児大脳型の発症年齢から2歳まで、それ以降ならできるだけ早期に診断することが大切です。一方、女性保因者では発症が成人以降になるため、成人、または児の出産を計画する前が推奨されています。
いずれの場合も診断する前に、専門家によるALDの病気についての詳しい説明を受けて理解することと、遺伝カウンセリングが必要です。
11.大脳型を発症する前に移植することはどうですか?
大脳型では、できるだけ早期の移植が推奨されています。しかし、大脳型発症前の移植は、その症例で大脳型を発症するかどうかその時点ではまだ判断できず、かつ移植のリスクもあるため、現時点では定期的な診察、脳MRI検査にて大脳型発症を確認次第、速やかに移植することが推奨されています。
12.遺伝子治療について教えてください。
2009年にフランスのグループが適切なドナーが得られなかった大脳型患者2例に、レンチウィルスを用いた遺伝子改変自己造血幹細胞移植を施行して、移植と同等の効果を得られたことを報告しました。現在、米国企業の支援にて海外で大脳型発症早期の患者に限定した治験が進行しています。2年経過時点で、遺伝子治療を受けた16例に移植関連死やGVHD(移植片対宿主病)は認めませんでしたが、1例に疾患進行死を認めています。他の15例は重篤な機能障害もなく生存しています。
13.新生児マススクリーニングについて教えてください。
米国では2013年末よりニューヨーク州において極長鎖脂肪酸を指標にした新生児マススクリーニングが開始され、3年間に70万人の新生児に対して45人のALD患者が発見されています。その後、2016年2月には米国におけるマススクリーニング対象疾患(RUSP)にALDが認定され、全米に広がりをみせています(ALD database )。
14.患者会について教えてください。
ALDの患者さんやご家族を支援する団体として、「ALD親の会」から発展したNPO法人「ALDの未来を考える会(A-Future)」があります。患者さんとそのご家族のQOL(生活の質)向上のための情報の収集と提供、介護サポート・心理ケア等の各種支援を目的としています。詳細はALDの未来を考える会(A-Future)のホームページをご覧願います。
特定非営利活動法人ALDの未来を考える会(A-Future)

15.公的支援について教えてください。
ALDの患者さんの公的支援としては基本的に18歳未満を対象とした小児慢性特定疾患と、年齢制限のない指定難病、両方とも対象疾患に認定されています。国内におけるALD患者さんの実情を把握する上でも重要な取り組みになりますので、申請につきましては主治医の先生にご相談願います。
詳細は下記のホームページをご覧願います。
指定難病20. 副腎白質ジストロフィー
小児慢性特定疾患104. 副腎白質ジストロフィー
16.患者登録制度について教えてください。
先天代謝異常症の患者登録制度としてJaSMIn(ジャスミン)があります。疾患名や現在受けている治療等といった患者さんの臨床情報を、患者さんご自身、あるいは保護者が登録していただく自己登録システムです。国内の希少疾患である先天代謝異常症の患者さんの情報を把握して、治療等に繋がる研究に寄与するとともに、患者さんに有益な情報を提供します。ALDの患者さんもこの制度に登録が可能で、ペルオキシソーム病に分類されています。
【参考】副腎白質ジストロフィー(ALD)診療ガイドブック2017

全文PDFは以下からダウンロードできます。
JaSMIn通信特別記事No.12(下澤先生)