JaSMInJapan Registration System for Metabolic & Inherited Diseases


ムコリピドーシス
国立成育医療研究センター遺伝診療科
小須賀 基通
1.はじめに
先天代謝異常症のなかで、ライソゾーム病と呼ばれる病気のグループには70種類以上の病気があると言われています。その中のひとつにムコリピドーシスという病気があります。ムコリピドーシスという名前は、この病気が臨床的にムコ多糖症とリピドーシス(脂質代謝異常症)の中間の症状を呈する病気として認識されたため、作られました。ムコリピドーシスは、ムコ多糖症とよく似た病気ですが、患者さんの数はさらに少なく、診断法もムコ多糖症とは違っています。今回はムコリピドーシスについて解説します。
2.原因
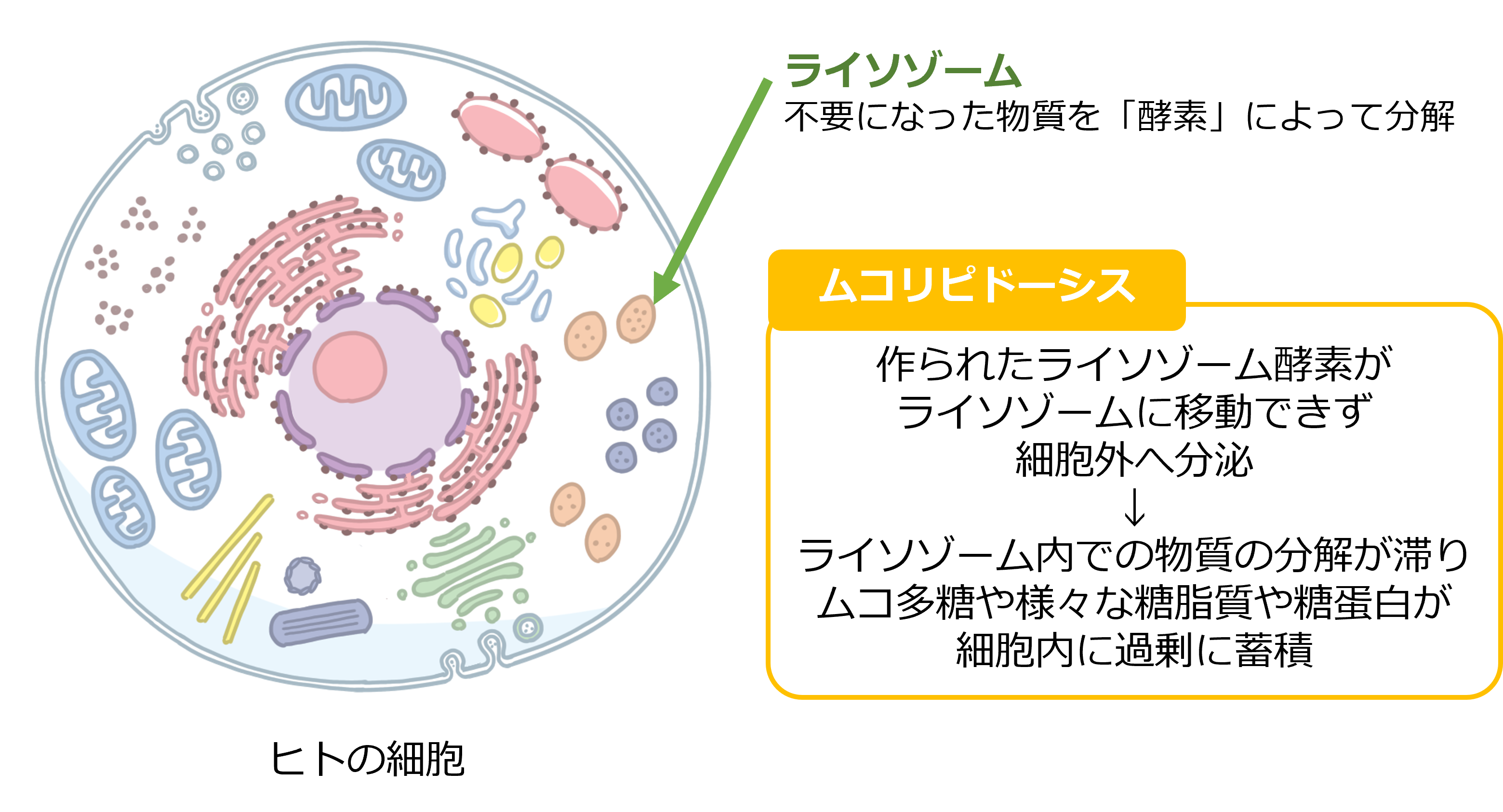
ヒトの細胞の中にあるライソゾームという器官は、細胞内で不要になったり古くなったりした物質を分解する働きがあります。様々な物質を分解するためにライソゾームには数十種類のライソゾーム酵素があると言われています。また細胞内で作られたライソゾーム酵素を効率良くライソゾームに移動させるためには、うまく取り込めるように目印のマーカーを付ける必要があります。一般的にライソゾーム病では、これらのうちの一つの酵素が生まれつき作ることができない、あるいは酵素の機能が働かないことが病気の原因となります。ムコリピドーシスでは、ライソゾーム酵素を作ることはできますが、そのマーカーを付けるために必要な酵素が働かないため、作られたライソゾーム酵素はライソゾームに移動できず、細胞外へ分泌されてしまいます。その結果、ライソゾーム内での物質の分解が滞り、ムコ多糖や様々な糖脂質や糖蛋白が細胞内に過剰に蓄積します。またムコリピドーシスII型をアイセル(I-cell)病とも呼びますが、アイセルとはinclusion cells(封入細胞)を略したものです。この病気の患者さんの皮膚の細胞を顕微鏡で見たときに、細胞の中のライソゾームが腫大して、inclusion(封入物)が見られたため、この細胞をinclusion cellsと呼び、さらにこれを略してI-cellとなりました。日本人でのムコリピドーシスの罹患率は、過去の報告では252,500人に1人であると推定されています。
3.症状
ムコリピドーシスの症状は、ムコ多糖症I型の重症型とよく似ています。すなわち、乳児期から角膜混濁、特徴的な顔つき、知的発達の遅れ、繰り返す難治性の中耳炎、難聴、肝臓・脾臓の腫大、関節の拘縮、骨の変形などを認め、これらは徐々に進行し、明らかになっていきます。以前は、乳児期に発症し症状の進行が早い重症型をムコリピドーシスII型、症状が軽く主に関節や骨の症状のみが見られ、ゆっくりとした経過をたどるものをムコリピドーシスIII型としていましたが、両者の分類や定義が困難な例も存在します。
4.診断
ムコリピドーシスでは、リンパ球などの細胞中のライソゾーム酵素は異常値を示さず、一方血漿や血清などの細胞外液中では、ほとんどのライソゾーム酵素活性が正常の5から20倍以上に増加しています。したがって、細胞と血漿あるいは血清の酵素活性の両方を測定し、その差を証明することで診断します。またライソゾーム酵素に目印のマーカーを付けるために必要な酵素に関係する遺伝子の異常を見つけることでも診断は可能です(遺伝子検査)。また臨床所見はムコ多糖症と似ていますが、ムコ多糖症のように尿中ムコ多糖(ウロン酸)の上昇は認めません。
5.治療
ムコリピドーシスには、まだ確立した有効な治療法はありません。ムコリピドーシスでは、ある特定の酵素のみの異常ではなく、すべてのライソゾーム酵素の転送障害が原因であるため、ムコ多糖症や他のライソゾーム病と違って、特定の酵素のみを酵素補充するというアプローチをとることはできません。過去には、骨髄移植、造血幹細胞移植が行われ、一部の患者さんには効果が見られましたが、現在では標準的な治療法と見なされていません。したがって、主な治療は、個々の症状に対する対症療法、リハビリテーション、療育などとなります。

全文PDFは以下よりダウンロードできます。